Exploratory data analysis of breast cancer transcriptomes
M Hallett
21/07/2020
Plan for Today
A few minutes about pipes in R (the \({\tt magrittr}\) package, also a part of \({\tt R}\)’s \({\tt tidyverse}\)).
Further explore our breast cancer transcriptome dataset. Our focus today is on the model in the Data Science cycle.
Here we focus more on the gene expression (rather than clinico-pathlogical sample data).
This will complete Part I of the textbook.
Today’s material marks the end of material covered by the first midterm.
Pipes
Pipes simplify R. They are a bit tricky at first.
The \({\tt tidyverse}\) incorporates the \({\tt magrittr}\) package. So when you load the \({\tt tidyverse}\) library, you have access to pipes.

To be honest, I think many new programmers can get confused by pipes so I would normally not teach them this early. However, the book and examples online do use pipes. So I guess we have to go with the flow.
Functions and composition
Approach 3 [Level Padawan] (composition of functions)
x <- 5
answer <- log(sqrt(exp(tanh(x))))Approach 4 [Level Jedi Warrior] (Pipes)
x <- 5
x %>% tanh %>% exp %>% sqrt %>% log -> answerHere \(x\) is transformed by a succession of functions. By default the left-hand side (LHS) will be piped in as the first argument of the function appearing on the right-hand side (RHS).
If it’s not the first argument, you can use “.” as a placeholder.
10 %>% seq(1, ., 2); 3 %>% seq(1, 10, .)
## [1] 1 3 5 7 9
## [1] 1 4 7 10
5 %>% seq( from = 1, by = 1) # fills in the missing parameter
## [1] 1 2 3 4 5Returning to our breast cancer transcriptome
We’ve learnt how to winnow our dataset down to the core that we want through \({\tt dplyr}\).
And we’ve learnt how to visualize that data to discover trends through \({\tt ggplot}\).
Now we focus on the types of questions we can ask, how to model our data (to answer these questions), and then to refine our data and visualizations (cyclic).
Modelling is a very big topic and one could argue that the a large portion of the course explores modelling from different angles.
Values and variability
brca <- rename(small_brca, stage = ajcc_pathologic_tumor_stage ) # just to simplify like last class
brca <- select(brca, -c("tss", "barcode", "id", "bcr_patient_uuid", "form_completion_date", "birth_days_to"))
ggplot(data = brca) + geom_bar(mapping = aes(x = stage)) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))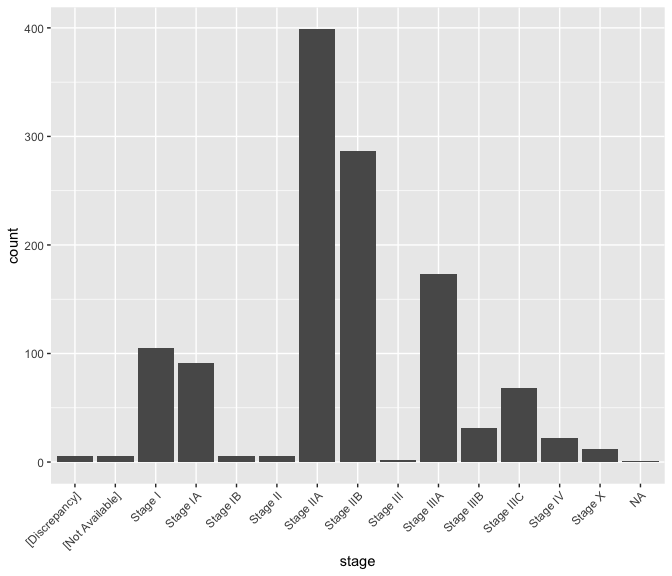
Explore other variables
ggarrange(g_gender, g_race, g_ethnicity, g_menopause_status,
ncol = 2, nrow = 2)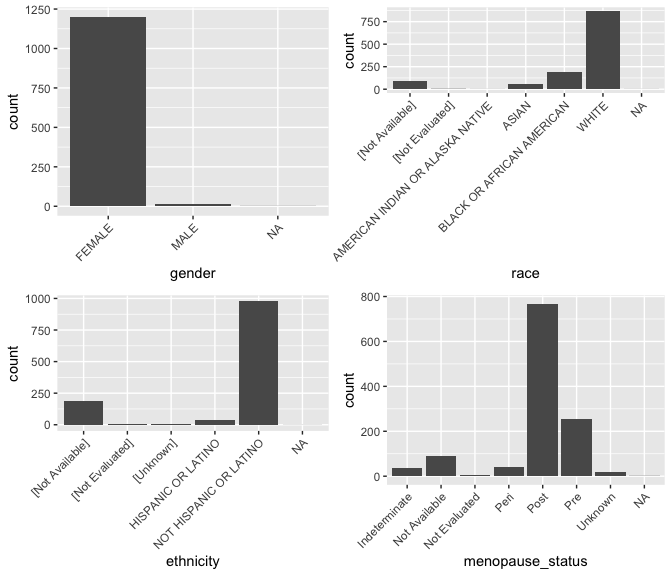
Explore other variables
ggarrange(g_hist, g_age, g_micromet, g_lymph,
ncol = 2, nrow = 2)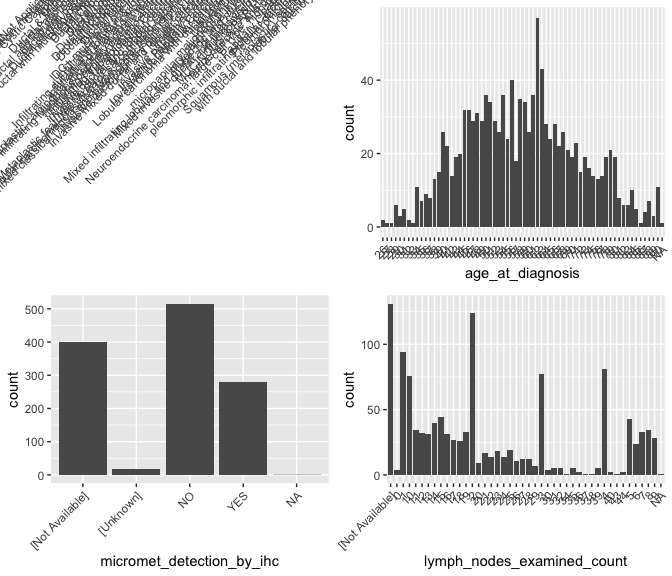
Explore other variables
ggarrange(g_er, g_pr, g_her2, g_mets, ncol = 2, nrow = 2)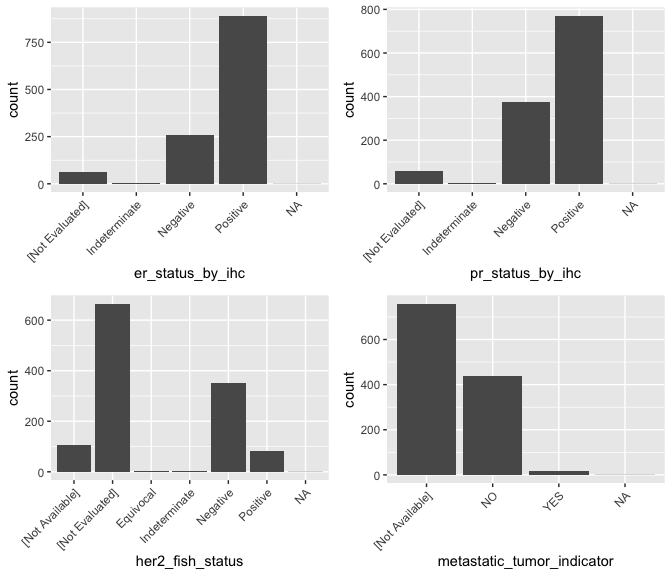
Histogram for gene expression
g <- ggplot(data = brca, mapping=aes(x = ESR1))
g1 <- g + geom_histogram() # I think default is 30 bins
g2 <- g + geom_histogram(bins=100)
g3 <- g + geom_histogram(bins=10)
g4 <- g + geom_histogram() + coord_cartesian(ylim = c(0, 500))
ggarrange(g1, g2, g3, g4, ncol = 2, nrow = 2)
## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.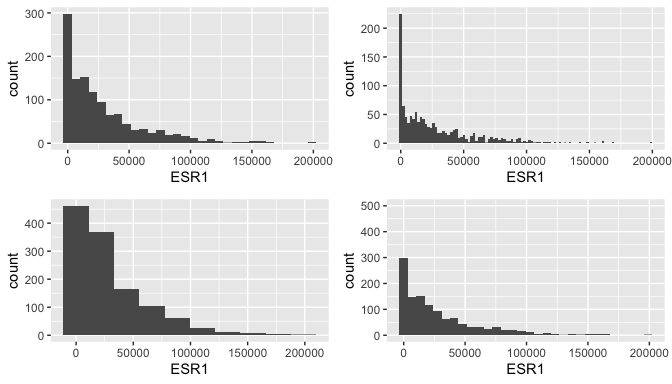
Box plots, the dinosaurs of visualization
- Displays the median (\(50^{th}\) percentile), the lower and upper hinge (\(25^{th}\) and \(75^{th}\) percentiles), two whiskers (about a \(95\%\) confidence interval from the tip of the lower whisker to the tip of the upper whisker), and outliers.
brca %>%
select( c(participant, tumor, "ESR1", "PGR", "ERBB2", "EGFR", "KRT5" )) %>%
melt(id.vars = c("participant", "tumor"), measure.vars = c("ESR1", "PGR", "ERBB2", "EGFR", "KRT5" )) %>%
as_tibble -> lava
ggplot(lava, aes( x= variable, y = value)) + geom_boxplot( ) + theme(axis.text.x = element_text(angle=65, vjust=0.6)) 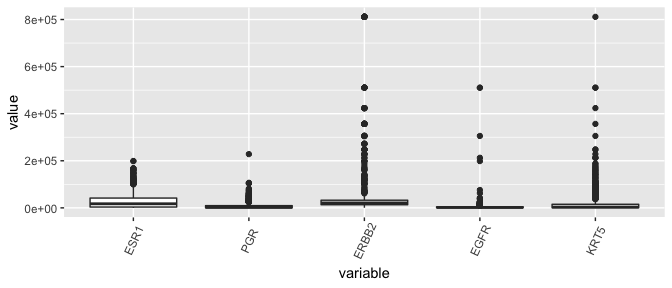
Yikes, this plot needs a \(log\)-transform as much as the entire population needs a haircut after the lockdown.
Log-transform before plotting
- When data spans multiple orders of magnitude, such as in this case where counts range from \(10^1\) to \(10^5\), it is important to \(log\)-transform data. That is, we compute \(log(x_y)\) where \(x\) is the count for gene \(y\).
ggplot(lava, aes( x= variable, y = value)) + geom_boxplot( ) +
scale_y_continuous(trans='log') + theme(axis.text.x = element_text(angle=65, vjust=0.6))
## Warning: Transformation introduced infinite values in continuous y-axis
## Warning: Removed 3 rows containing non-finite values (stat_boxplot).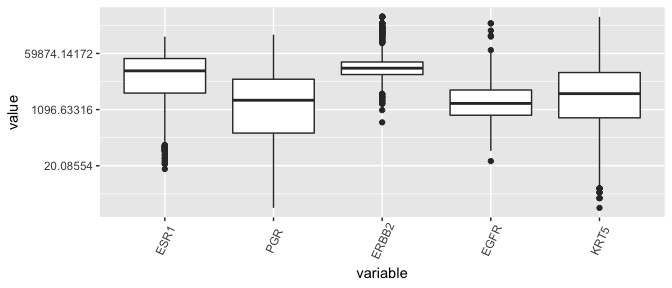
Violin plots: First \(10\) genes, tumor vs normal
- Violin plots killed the box plot (like video killed the radio star).
brca %>% select( c(participant, tumor, ANLN:ESR1 )) %>%
melt(id.vars = c("participant", "tumor"), measure.vars = colnames(brca)[24:33]) %>%
as_tibble -> lava
ggplot(lava, aes( x= variable, y = value, fill=tumor)) +
geom_violin( trim = FALSE, position = position_dodge(0.9), draw_quantiles = c(0.5) ) + scale_y_continuous(trans='log') +
theme(axis.text.x = element_text(angle=65, vjust=0.6)) 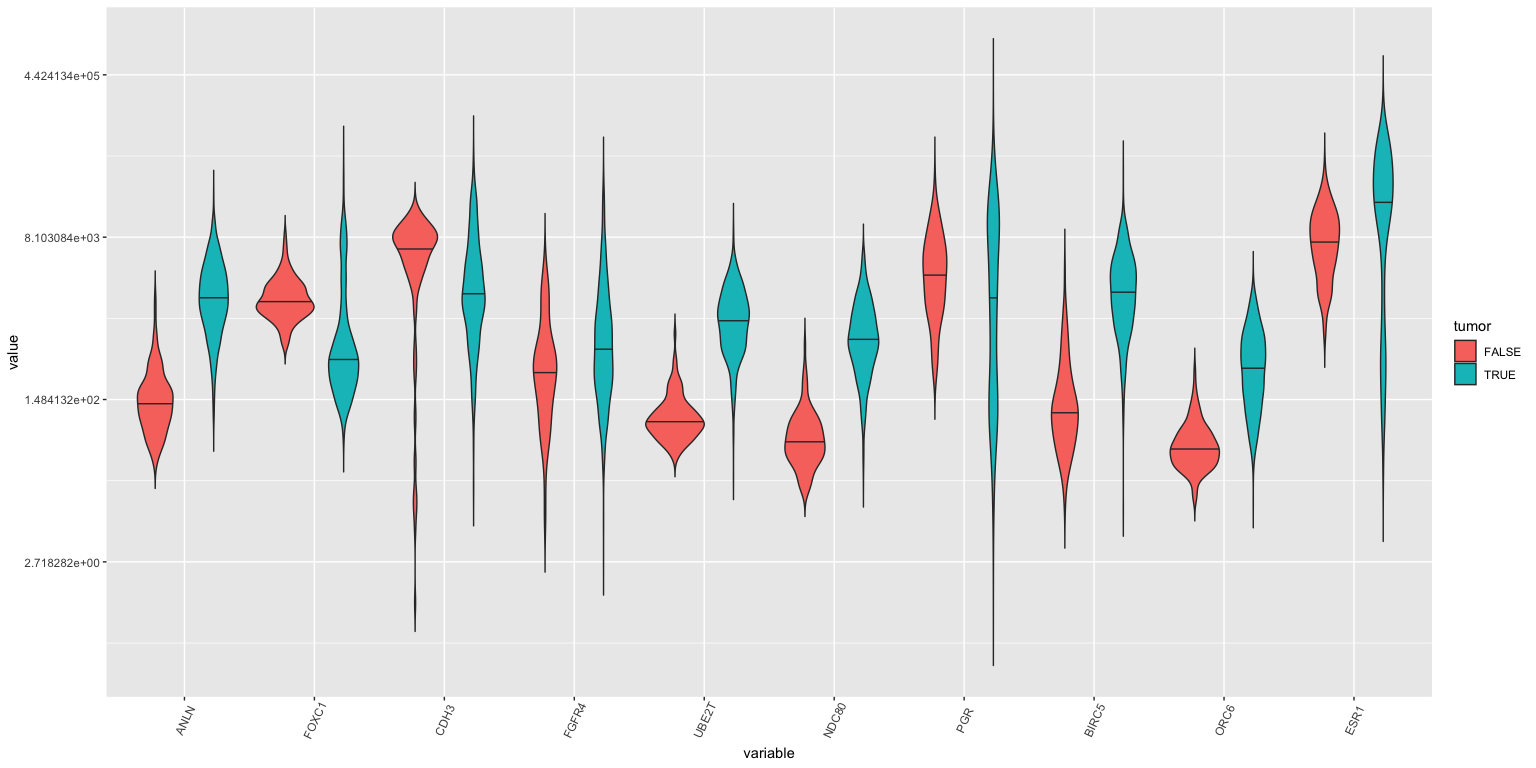
Violin plots: All genes, tumor vs normal
## Warning: Transformation introduced infinite values in continuous y-axis
## Warning: Removed 90 rows containing non-finite values (stat_ydensity).
## Warning in regularize.values(x, y, ties, missing(ties), na.rm = na.rm):
## collapsing to unique 'x' values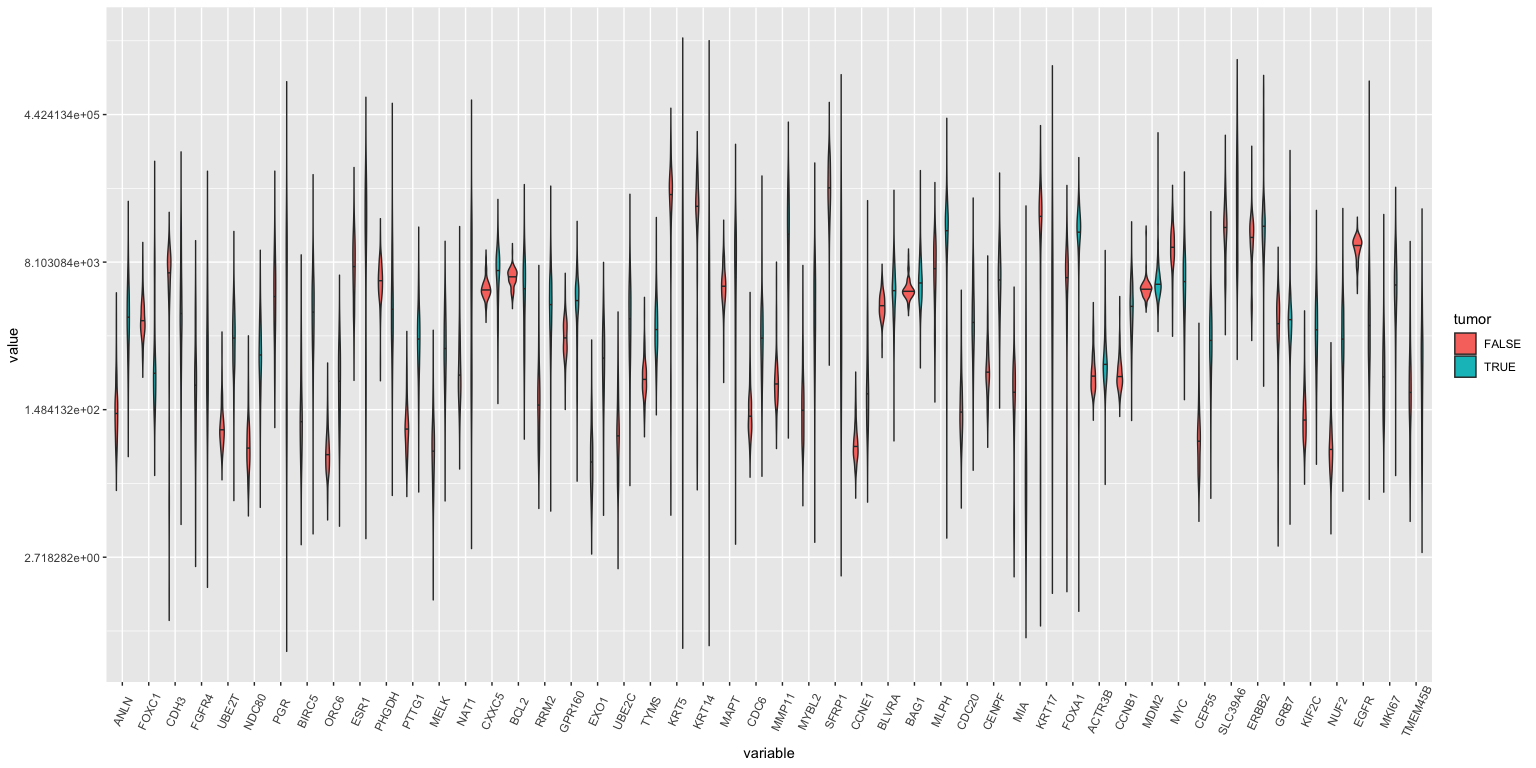
If you study this closely (with a magnifying glass), you’ll see that ANLN, UBE2T, ORC6 amongst others are different between tumor and normal samples.
Modelling tissue status using gene expression
- Could we use the expression levels of ANLN and UBE2T to differentiate between healthy breast and tumor material? I’m not sure what the clinical utility of this would be ultimately, but it serves as a simple example to start. We will tackle more complicated questinos later.
ggplot(brca, aes(x=ANLN, y=UBE2T, color=tumor), size = .3) +
scale_y_continuous(trans='log') + scale_x_continuous(trans='log') +
geom_point() + geom_rug()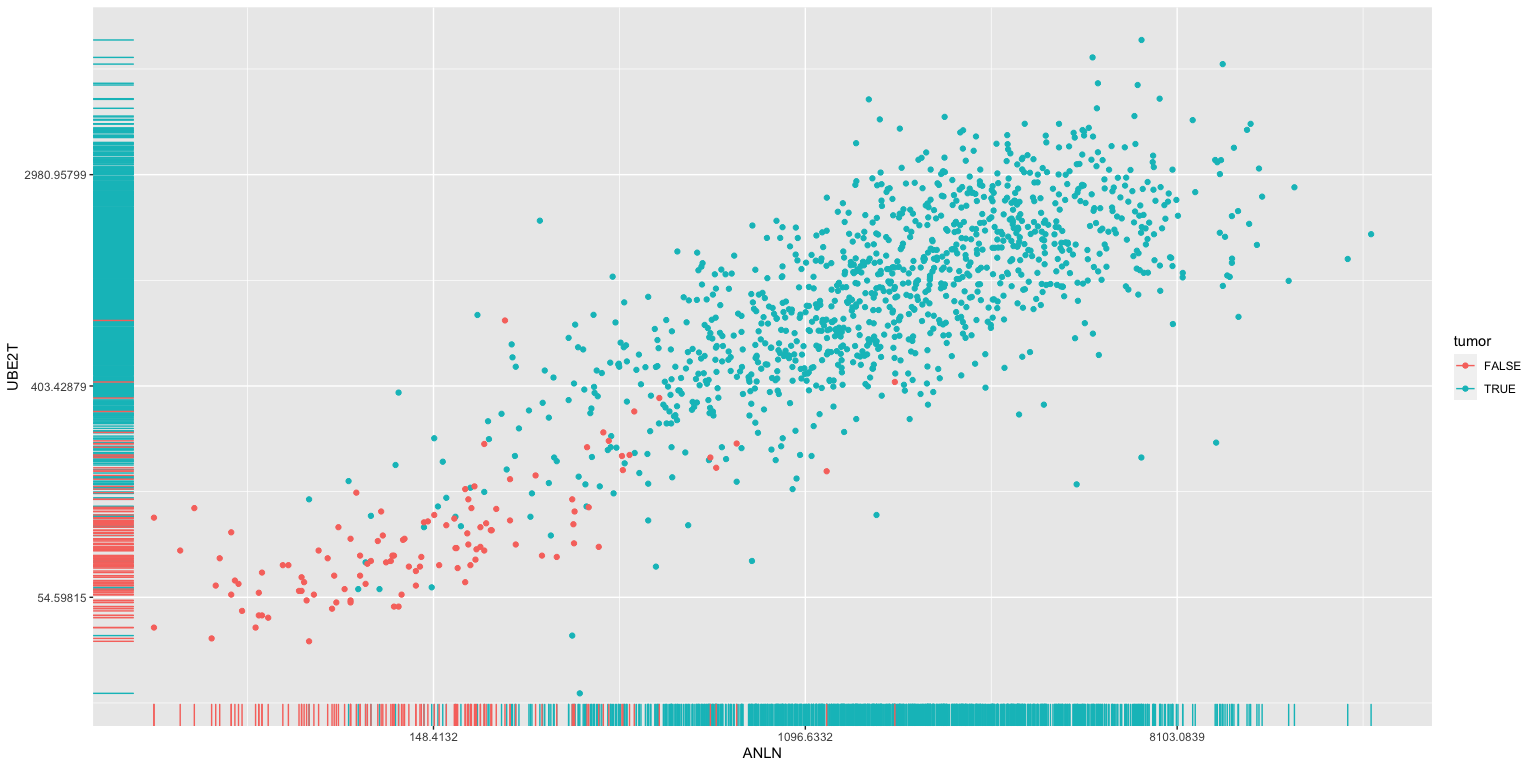
BIOL480
© M Hallett, 2020 Concordia University