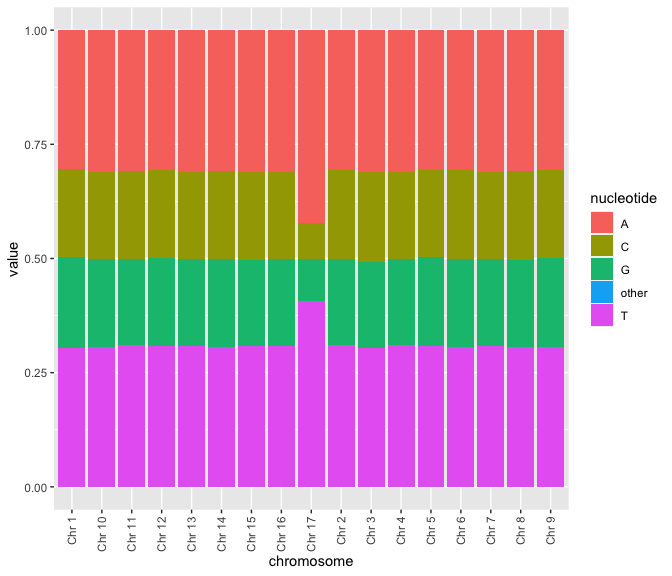
Not surprisingly, genomes are commonly required for analysis (otherwise, why would we be doing all this sequencing?)
Some are small (viruses) and some are big (plants). R Shiny app
Some are circular including virsuses, prokaryotes (that is, Bacteria and Archae/without nucleus) and mitochondria of Eukaryota.
Some are linear (Eukaryota but some bacteria have linear genomes).
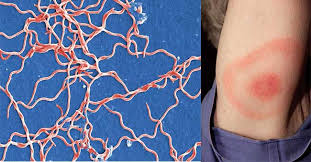
Borrelia burgdorferi; what disease is this?
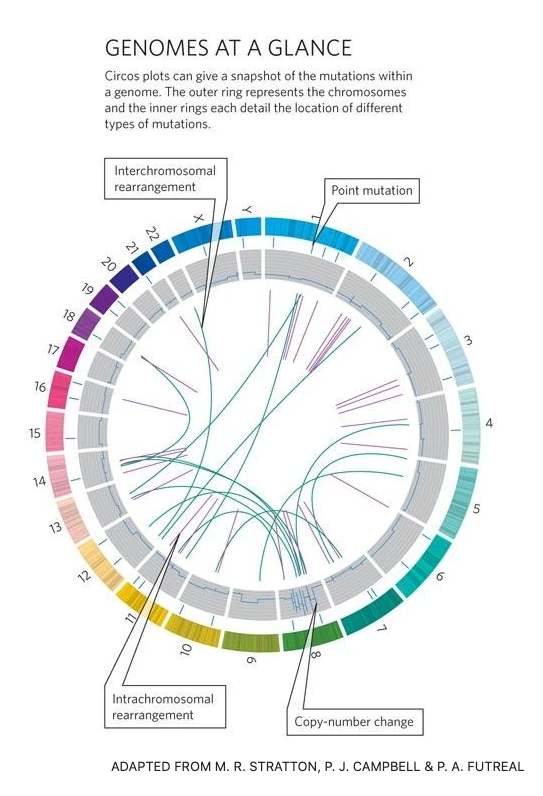
Cancer genomes
A hallmark of cancer is genomic instability.
The neoplasticity of cancer genomes results in novel point mutations, chromosomal amplifications, loss, loss of heterozygosity, aneuploidies, euploidies etc.
Circos plot comparing cancer versus normal genome
Genomes
How many genomes have been sequenced to date? It depends a bit on how you count.
(Except for IGV, all of these efforts also act as a repository for genomes. NCBI is most complete but UCSC and Ensembl have most model organisms.)
What are the issues here?
What if you want to ask the question of the form: What binding sites are in the promoter of gene X? Where are the enhancers for gene Y? How do transcription start sites change across the genome? Where are all the cut sites for a restriction enzyme? For a region R on chromosome C of organism X, what is its syntenic regions across all fungi?
For these kinds of questions, visualization tools won’t suffice.
For that we need the data represented in good data structures and the capcity to manipulate it
Where we are and what is required
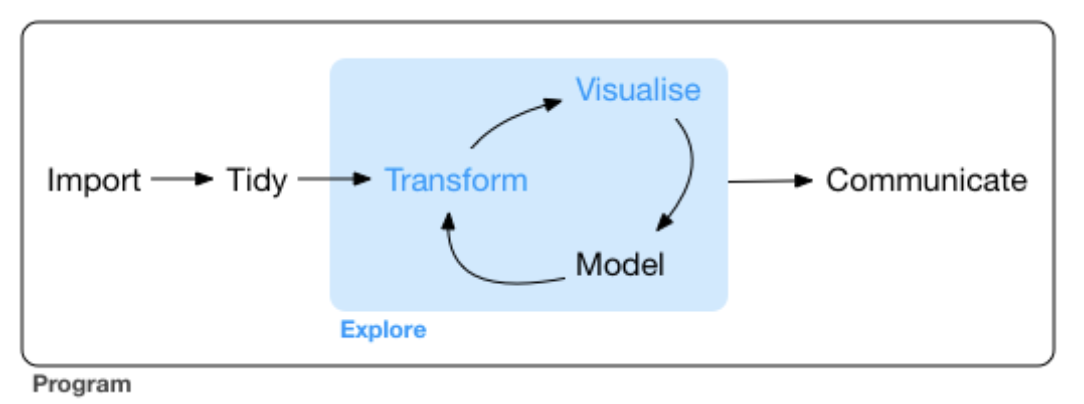
From R for Datas Sience, Chapter 6.
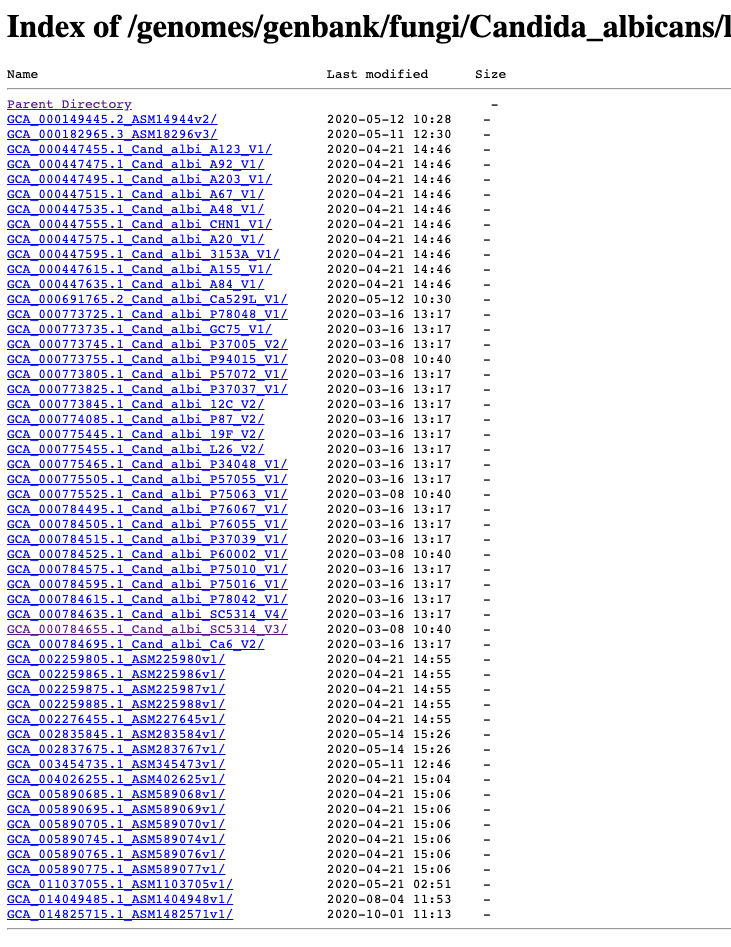
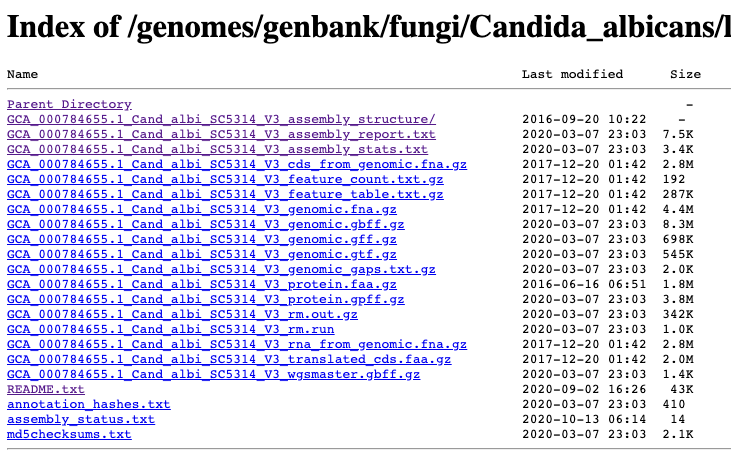
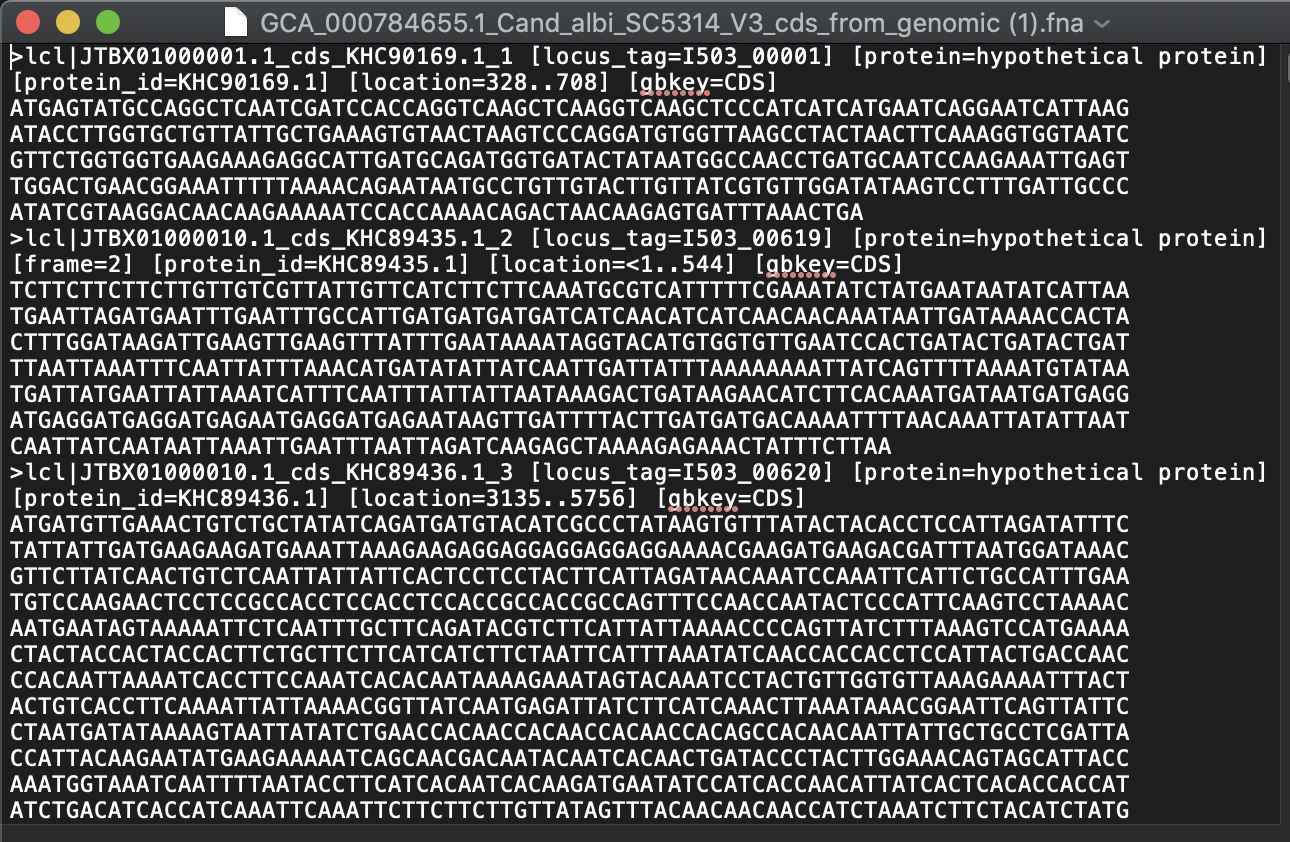
Where to get the genomes and annotations from?
How to represent them in R? (not tidy data but other packages)
A quick note about different versions of the human genome
There are at least three major versions of the human reference genome. One from the Genome Reference Consortium, one from the UCSC genome browser database and one from the Broad Institute.
Why? Mostly the same I believe but differ slightly on a few N bases (undetermined) and how the mitochondrial genome is reported.
It is also possible to download genomes and annotations directly into R from the UCSC Genome Browser effort.
The \({\tt BSGenome}\) packages from Bioconductor are available for many organisms here.
The data is uploaded into a \({\tt Biostrings}\) object (more in a bit).
Back in the days… characters
In base R (the traditional version of the software), we had the \({\tt character}\) class.
x <- "The rockets are distributing about London just as Poisson’s equation in the textbooks predicts"nchar(x); substr(x, start=5, stop =11);
## [1] 94
## [1] "rockets"
y <- "As the data keep coming in, Roger looks more and more like a prophet."toupper(y) # not to mention tolower
## [1] "AS THE DATA KEEP COMING IN, ROGER LOOKS MORE AND MORE LIKE A PROPHET."
Back in the days… characters
z <-cat(x, y); z # careful
## The rockets are distributing about London just as Poisson’s equation in the textbooks predicts As the data keep coming in, Roger looks more and more like a prophet.
## NULL
z <-paste(x, y, sep =". ")gregexpr(pattern="on", text = z)
## [1] "The rockets are distributing about London just as Poisson’s equation in the textbooks predicts. As the data keep coming in, Mike looks more and more like a prophet."
And then the tidyverse: stringr
It’s nicer.
str_length( c(x, y))
## [1] 94 69
z <-str_c(x, y, sep=". ")pizza <-TRUEnum_kids <-3str_c( "Ok my ", num_kids, " spawn, we should", if (pizza) " order pizza."else" eat lettuce." )
## [1] "Ok my 3 spawn, we should order pizza."
stringr
str_sub( x, start=5, end=11)## [1] "rockets"str_to_upper(x)## [1] "THE ROCKETS ARE DISTRIBUTING ABOUT LONDON JUST AS POISSON’S EQUATION IN THE TEXTBOOKS PREDICTS"str_sort( c("one", "two", "three", "four"))## [1] "four" "one" "three" "two"# str_
stringr
str_view(c(x,y), "o.e")
str_view(c(x,y), "..$")
str_view(z, "keep(s|)")
Biostrings
Base R and stringr functions are for small short strings.
Genomic data is much bigger and requires special data structures to efficiently store them.
Also, genomic data requires functions especially designed to nucleic and amino acid sequences (eg alignments which we will talk much more about over the next few weeks).
\({\tt Biostrings}\) is an important package for manipulating genomic strings.
Biostrings
Biostrings allows for DNA strings (DNAString), RNA strings (RNAString), and amino acid strings (AAString) with generic classes BString and XString (more about these last two later).
# BiocManager::install("Biostrings")library(Biostrings)dnastring =DNAString("GCGATN-CTC")dnastring## 10-letter DNAString object## seq: GCGATN-CTClength(dnastring)## [1] 10DNA_ALPHABET## [1] "A" "C" "G" "T" "M" "R" "W" "S" "Y" "K" "V" "H" "D" "B" "N" "-" "+" "."alphabetFrequency(dnastring, baseOnly=TRUE, as.prob=TRUE)## A C G T other ## 0.1 0.3 0.2 0.2 0.2
An important concept in computational biology is the notion of an alignment: how to find one string within another string.
PpiI <- "GAACNNNNNCTC"# a restriction enzyme pattern(align.PpiI <-matchPattern(PpiI, phiX174Phage[[1]], fixed=FALSE))
## Views on a 5386-letter DNAString subject
## subject: GAGTTTTATCGCTTCCATGACGCAGAAGTTAACA...CTTCGATAAAAATGATTGGCGTATCCAACCTGCA
## views: NONE
Biostrings
The previous slide explored a very simple notion of alignment, basically an “exact” match. There are more biologically relevant notions of alignment that ultimately are based in models of evolution. We will learn these in part 3 of the course.